

In the US and the EU, the requirements for medical device labeling are detailed and extensive and may be specific to the type of device. Manufacturers should always refer directly to the text of the applicable laws to ensure that they are following the correct procedures and staying compliant. A label is any “display of […]
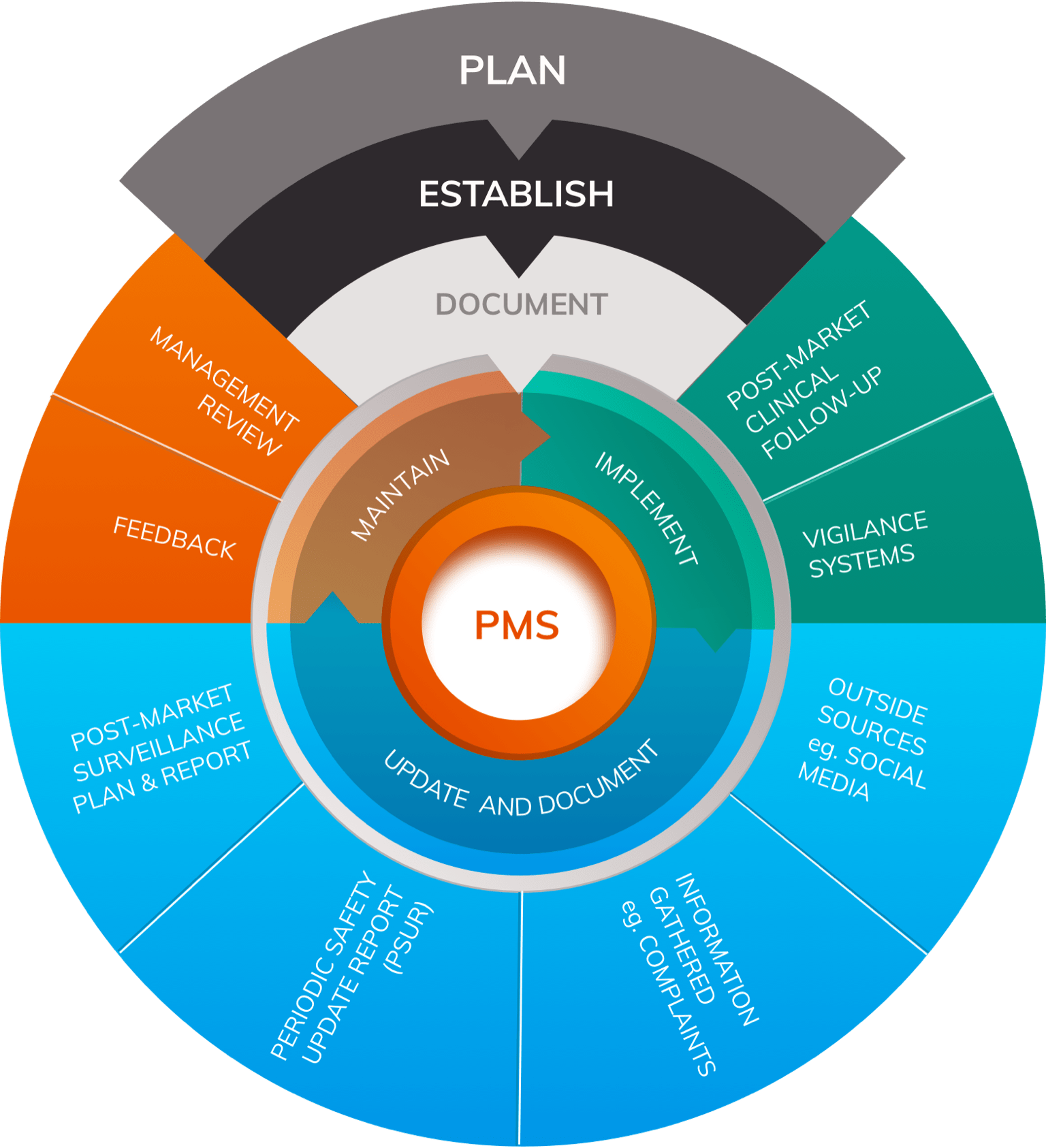
Post Market Surveillance is undertaken as a responsibility of the manufacturer it is different from market surveillance which is used to describe activities to monitor compliance with the Regulations undertaken by and coordinated between national competent authorities. 1. Class I devices are typically exempt from post market surveillance requirements 2. PMS need to carry out […]

Pre-Market Submission 510(k) must demonstrate that the device is substantially equivalent to one legally in commercial distribution in the United States: (1) before May 28, 1976; or (2) to a device that has been determined by FDA to be substantially equivalent. There are three different types of 510(k) submissions. a. Special 510(k): This type of […]

The Medical Device Single Audit Program (MDSAP) is a program that allows the conduct of a single regulatory audit of a medical device manufacturer’s quality management system that satisfies the requirements of multiple regulatory jurisdictions. Audits are conducted by Auditing Organizations (AO’s) authorized by the participating Regulatory Authorities to audit under MDSAP requirements. The MDSAP […]
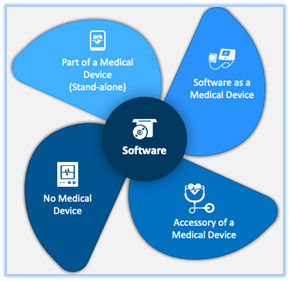
Software will dominate the medical device market in the near future as technology develops and becomes more and more embedded in digital platforms, where its applications in the healthcare and medical sectors are particularly prominent. The IMDFR defined SaMD as a “software intended to be used for one or more medical purposes that perform these […]
Hop on this transformational journey with us.
Contact Us