Digital Health
The categories of digital health include mobile health (mHealth), Health Information technology (IT), wearable devices, telemedicine, telehealth and personal medicine. Digital health has the potential to improve the ability to accurately diagnose and treat diseases which enhances the delivery of health care for the individual.
The applications of these technologies range from general wellness to applications as a medical device. They include technologies intended to use as a medical product, as a companion diagnostics or an adjunct to other medical products (devices, drugs and biologics). They are also used to develop or study medical products.
The technologies such as smart phones, social media and internet applications are only changing the way we communicate; it is also providing us with ingenious paths to monitor the health and greater access to information.
Benefits of Digital health technologies
The most important benefit of digital health technology to the patient is the accessibility of data and thereby giving more control for patients over their health. It gives real opportunities to improve their health which enhances efficiency.
These technologies can empower patients to make better decisions about their health and opens up new options for early diagnosis of life threatening diseases and other also better management of chronic conditions.
Following are the benefits expected by providers and other stakeholders when these technologies used.
– Reduced inefficiencies,
– Improved access,
– Reduction in costs,
– Increase in quality, and
– Personalized medicine for patients.
For patients and consumers, these technologies help in better management and tracking of their health and wellness related activities.
FDA’s Focus on Digital Health technologies
Modern medical devices have the ability to connect and communicate with other systems. Meanwhile, devices that are already approved are being upgraded to add digital features. New categories of devices are being explored.
Following topics are the point of interest for FDA which is trying to provide more clarity using practical approaches that balance benefits and risks.
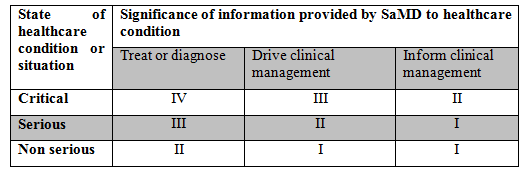
– Software as a Medical Device (SaMD)
SaMD is defined as ‘software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device’.
This can be used in various platforms such as medical device platforms, commercial off the shelf platforms, virtual networks etc. these systems were previously known as ‘Stand alone software’, ‘medical device software’, or ‘health software’.
– Artificial Intelligence and Machine Learning (AI/ML) in Software as a Medical Device
Artificial Intelligence (AI) is broadly defined as producing intelligent machines, especially computer programs with the help of science and engineering. AI uses varied techniques such as statistical analysis data, machine learning and so on.
Machine Learning (ML) is one of the AI technique used to design and train software algorithms based on the obtained data. Software developers can make use of ML to create an algorithm which can be ‘Locked’ or ‘Adaptive’. These two decides the behavior of the device when new data is gathered.
Since AI and ML technologies have immense potential to transform healthcare by providing the important insights based on the data gathered every day. The FDA’s CDRH is considering a regulatory framework which should include total product life cycle which will allow modifications to be made from real world learning adaptation while ensuring the safety and effectiveness of the device.
– Cyber security
In the present scenario, there is an increase in the cyber security risks to medical devices as they are connected to internet and other networks which can be easily hacked if there is no efficient security which impacts the safety and effectiveness of the device. The healthcare environment is complex, all the stakeholders must work together to ensure the cyber security risks are mitigated and eliminated time to time.
– Device Software Functions, including Mobile Medical Applications
The software functions that meet the definition of a device are deployed on mobile platforms, general purpose computing platforms, or in the function or control of a hardware device.
Mobile apps are primarily software programs that run on smart phones and other mobile devices. They can also be used as accessories that attach to a smart phone or other devices or combination of accessories and software. Mobile apps can help people manage their own health and wellness there by promoting healthy living, and easy access to information.
The FDA encourages the development of Mobile Medical Apps (MMAs) that improve the health and provide both consumers and health care professionals with valuable health information. FDA also has responsibility to oversee the safety and effectiveness of medical devices including MMAs. The FDA applies the same risk based approach to device software functions as other medical devices.
For many software functions that meet the regulatory definition of a ‘device’ but poses minimal risk. For these devices, FDA will not expect manufacturers to submit premarket review application or to register and list their software with the FDA.
– Health IT
Health Information technology or health IT is defined as ‘Hardware, Software, Integrated technologies or related licenses, Intellectual property, upgrades, or packaged solutions sold as entities or patients for the electronic creation, maintenance, access or exchange of health information.
FDA worked with Federal Communications Commission (FCC), and National coordinator for heath IT (ONC) to propose a strategy and make recommendations on appropriate risk based regulatory framework for health IT that promotes innovation, protects patient safety and avoids unnecessary and duplicate regulation.
The three agencies are committed to a vision that supports a strong health system based on safe and innovative health IT that improves and advances public health.
On 4/3/2014, the FDA, FCC and ONC released the Food and Drug Administration Safety and Innovation Act (FDASIA) Health IT Report outlining a proposed strategy and recommendations for a risk-based framework.
– Medical Device Data Systems
Medical Device Data Systems (MDDS) are hardware or software products intended to transfer, store, convert formats, and display medical device data. A MDDS does not modify the data or modify the display of the data, and it does not by itself control the functions or parameters of any other medical device. MDDS may or may not be intended for active patient monitoring.
– Medical Device Interoperability
Medical device interoperability is the ability to safely, securely, and effectively exchange and use information among one or more devices, products, technologies, or systems. This exchanged information can be used in a variety of ways including display, store, interpret, analyze, and automatically act on or control another product.
Interoperable devices with the ability to share information across systems and platforms can:
– Improve patient care,
– Reduce errors and adverse events,
– Encourage innovation, and
– Enable more diverse study datasets.
– Telemedicine
The Health Resources and Services Administration (HRSA) define telehealth as ‘the use of electronic information and telecommunications technologies to support and promote long-distance clinical health care, patient and professional health-related education, public health and health administration’. Technologies which fall under this category are videoconferencing, the internet, store-and-forward imaging, streaming media, and terrestrial and wireless communications.
– Wireless Medical Devices
Radio frequency (RF) wireless medical devices perform at least one function that utilizes wireless RF communication such as Wi-Fi, Bluetooth, and cellular/mobile phone to support health care delivery. Examples include controlling and programming a medical device, monitoring patients remotely, or transferring patient data from the medical device to another platform such as a cell phone. As RF wireless technology continues to evolve, this technology will increasingly be incorporated into the design of medical devices.
The use of RF wireless technology can translate to advances in health care, and patients should be informed about the safe and effective use of these devices in the course of daily life.
Health care facilities should consider the following:
– Selection of wireless technology
– Quality of service
– Coexistence
– Security
– Electromagnetic Compatibility (EMC)
Digital Health Center of Excellence
For the advancement of digital health technologies, CDRH has established Digital Health Center of Excellence which helps the digital health stakeholders to advance health.
The aim of the Digital Health Center of Excellence is to
– Share knowledge
-Connect and build partnerships
– Innovate regulatory approaches
Through fulfilling the above objectives, the following advancements in digital health have been anticipated
– Advancement in the science of digital health that meets the requirements of stakeholders
– Well organised structure for the easier access to highly specialised expertise, information and tools to accelerate digital health technology
– Aligned regulatory approach to harmonize international regulatory expectations and industry standards
– Increased awareness and understanding of digital health trends
– Consistent application of digital health technology policy and oversight approaches
– Restructured medical device regulatory model specifically for digital health