

History:
The Medical Audit Single Audit Program (MDSAP) was developed by the International Medical Device Regulators Forum (IMDRF) to allow third party auditors to conduct a single audit of a medical manufacturer that covers ISO 13485:2016 and the respective regulatory requirements.
A working group was established for MDSAP in 2012. Then, IMDRF initiated a three year pilot program between 2014 and 2016.
The main goal of the MDASAP program is to establish a union internationally that can work together to provide oversight and safety on a global scale to the medical device manufacturers.
Participating partners:
International partners that are participating in the MDSAP include:
MDSAP Members:
a) Therapeutic Goods Administration of Australia (TGA)
b) Brazil’s Agência Nacional de Vigilância Sanitária (ANVISA)
c) Health Canada (HC)
d) Japan’s Ministry of Health, Labour and Welfare, and the Japanese Pharmaceuticals and Medical Devices Agency
e) U.S. Food and Drug Administration (USFDA)
MDSAP Official Observers:
a) European Union (EU)
b) United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA)
c) The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Programme
MDSAP Affiliate Members:
a) Argentina’s National Administration of Drugs, Foods and Medical Devices (ANMAT)
b) Ministry of Health of Israel (NEW)
c) Republic of Korea’s Ministry of Food and Drug Safety
d) Singapore’s Health Sciences Authority (HSA)
Status of MDSAP adoption in participating countries:
a) US: FDA will accept the results of MDSAP audits rather than conducting inspection. However, the agency still conducts its own initial visits to manufacturers and ‘for cause’ inspections.
b) Canada: after January 1st 2019, HC fully transitioned to accept only MDSAP compliance audits from the manufacturers.
c) Brazil: the authority will accept MDSAP for initial audits. However, it will continue to conduct its own inspections and audits for high risk devices.
d) Japan: the authority has fully adopted MDSAP and will accept MDSAP audit results in place of J-QMS audit.
e) Australia: another adopter of MDSAP. The authority recognizes manufacturers who pass the audit and satisfied their QMS requirements. The TGA recognizes MDSAP certificates as equivalent to CE certificates.
Eligibility:
a) Any manufacturer may participate in the audit program if their product falls under the scope of at least one participating country and subject to QMS requirements
b) Located anywhere in the world are eligible to participate
c) Only the MDSAP participating countries will have access to audit report
d) Manufacturers cannot select which of the 5 regulatory schemes to be included in the audit scope. All country specific requirements of the manufacturer’s target sale countries must be included
MDSAP actors:
Regulatory Authorities (RA): They are responsible for designating the Auditing Organizations (AO). There are certain criteria to be fulfilled to designate AO. The RA’s will continue to monitor the program and they have the final decision.
Auditing Organization (AO): They plan, conduct and report the audit to RA’s.
Manufacturer: they engage with the AO for QMS audits. If there is any non conformity, they need to provide corrective action plan.
Benefits of MDSAP certification:
-Reduces the burden for a manufacturer in case of audits and inspections
-Minimizes business obstacles, reduces cost and saves time due to the single audit process
-Helps the manufacturer to enter new markets with ease
-Consistent multiple regulatory programs by participating regulators
-Positive image for the company
MDSAP audit cycle:
The MDSAP audit program is based on three year audit cycle. The initial audit, also referred to as ‘Initial Certification Audit’ consists of stage 1 and stage 2 audit. The initial audit is followed by partial surveillance audit and a complete Re audit, also referred to as ‘Recertification audit’ in the third year. A recertification audit may include stage 1 audit, if there are any significant changes to the already audited QMS.
Special audits are extraordinary audits that are not part of the planned audit cycle. These audits mainly focus on specific elements of QMS. These include audits conducted in response to an application for the extension to the scope of an existing certification, to determine whether or not the extension can be granted or as short-notice audits conducted to investigate potentially significant complaints, or if specific information provides reasons to suspect serious non-conformities of the devices, or for other reasons.
Unannounced audits: The MDSAP participating regulatory authorities require AO to conduct unannounced audits in circumstances where high grade non-conformities have been detected.
MDSAP audit process:
The MDSAP audit process involves seven chapters or processes. They are, four primary processes, one enabling process and two supporting processes which are mentioned below.
-Management
-Measurement, Analysis and Improvement
-Design and Development
-Production and Service Controls
-Purchasing
-Device Marketing Authorization and Facility Registration, and
-Medical Device Adverse Events and Advisory Notices Reporting
Each chapter contains multiple audit tasks that are verified during the audit. Each audit task references the applicable clauses of ISO 13485:2016. Below table gives an overview of number of tasks mentioned in each chapter.
| MDSAP PROCESS | NUMBER OF TASKS |
| Management | 11 |
| Device Marketing Authorization and Facility Registration | 03 |
| Measurement, Analysis and Improvement | 16 |
| Medical Device Adverse Events and Advisory Notices Reporting | 02 |
| Design and Development | 17 |
| Production and Service Controls | 29 |
| Purchasing | 12 |
MDSAP audit sequence:
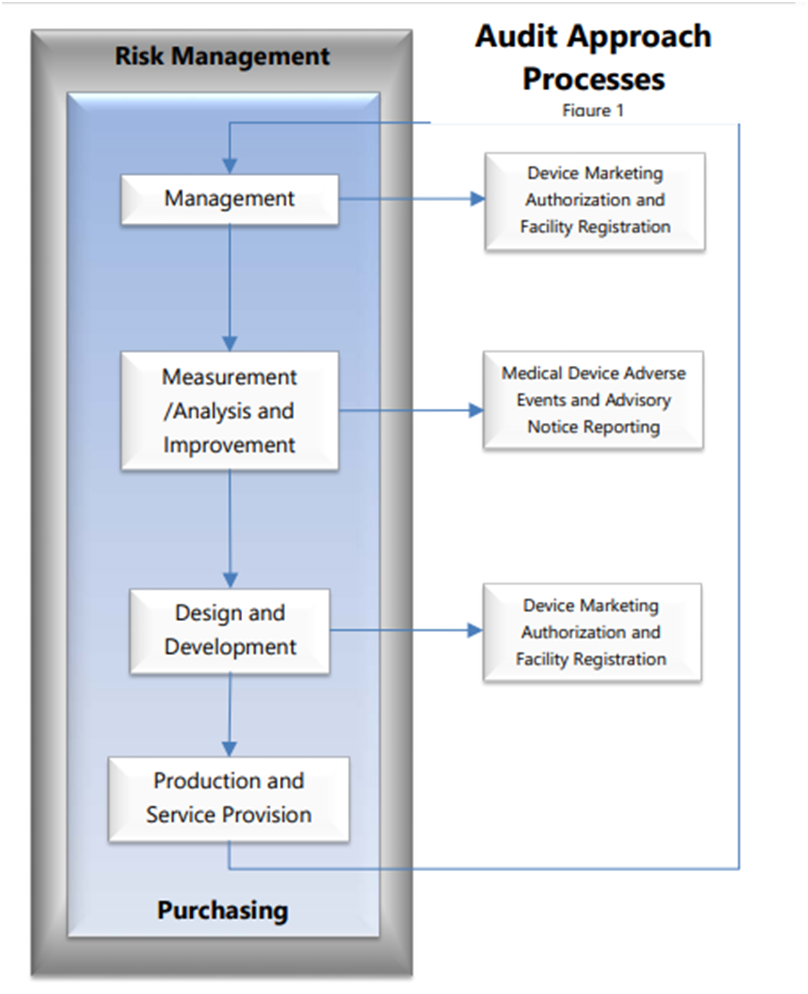
The MDSAP audit sequence follows a process approach and has four primary processes – Management process, Measurement, Analysis and Improvement process, Design and Development process and a Production and Service Controls process with links to the supporting process for Purchasing. Also, it has two additional supporting processes: Device Marketing Authorization and Facility Registration and Medical Device Adverse Events and Advisory Notices Reporting. These are necessary to fulfill specific requirements of the participating MDSAP regulatory authorities.
The audit sequence should be followed as designed. However, under certain circumstances, judicious exceptions to the audit sequence are allowed considering there is sufficient justification and the core elements of the MDSAP Audit Approach, are respected.
The flowchart shown in Figure 1 illustrates the MDSAP audit sequence and interrelationships.
MDSAP grading system:
The MDSAP grading system was created to clarify and address inconsistencies within traditional audit grading approaches that make use of ‘Major findings’ or ‘Opportunity for Improvement’.
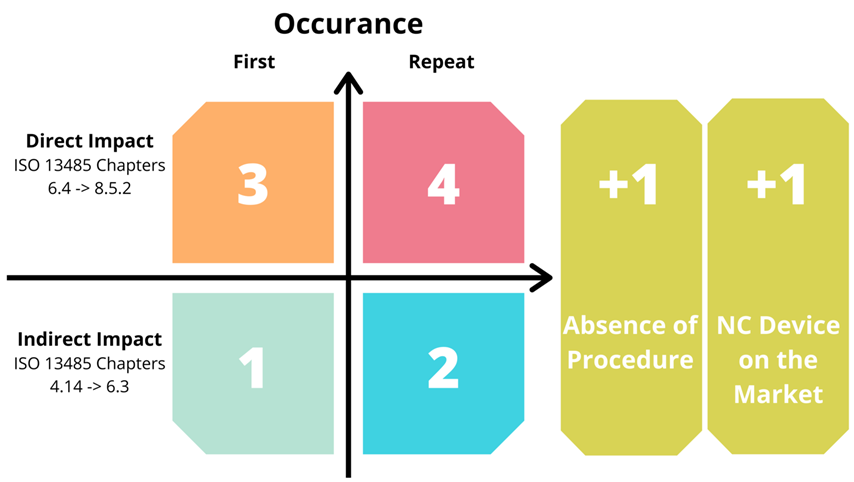
Non Conformities (NC) are assigned a grade between 1 and 5, which is calculated using a two step scoring system.
To determine the initial score, four point NC matrix is used. There are two categories based on clauses of ISO 13485:2016.
Indirect – Clauses 4.1 through 6.3, which have indirect impact on safety and performance of a medical device, it includes general documentation, and quality manual(s).
Direct – Clauses 6.4 through 8.5.3, which has direct impact on medical device safety and performance. The documents are mainly related to design, production, CAPAs etc.
Below table gives an insight in to the NC grading system.
Why choose KN consulting
KN consulting and services takes a proactive approach in informing our customers about the regulatory changes concerning the industry.
-Saves time and money
-Faster market access
-Expert guidance
