In the US and the EU, the requirements for medical device labeling are detailed and extensive and may be specific to the type of device. Manufacturers should always refer directly to the text of the applicable laws to ensure that they are following the correct procedures and staying compliant.
A label is any “display of written, printed, or graphic matter upon the immediate container of any article,” and must be clearly visible even if the “immediate container” has additional packaging. “Label” also includes anything printed on a product container or wrapper, or anything that accompanies the article when it is put up for sale. The label cannot include any false or misleading statements, and must be displayed prominently in an appropriate location
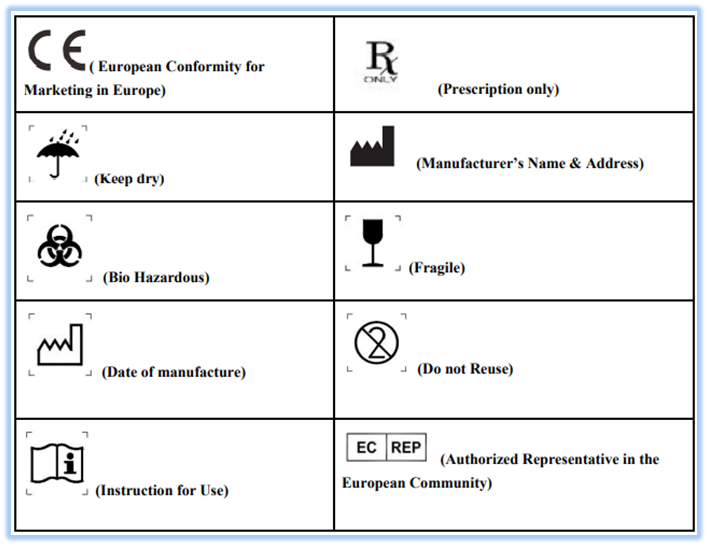
ISO 15223-1:2016
One of the important standards for marketing and this standard identifies requirements for symbols used in medical device labeling that convey information on the safe and effective use of medical devices. This standard contains all the required symbols for a medical device i.e. Reference number, Title of symbol, Description, Requirements, Informative notes, Restriction of use, Additional requirements and Registration number. Examples for commonly used labels are as follows: