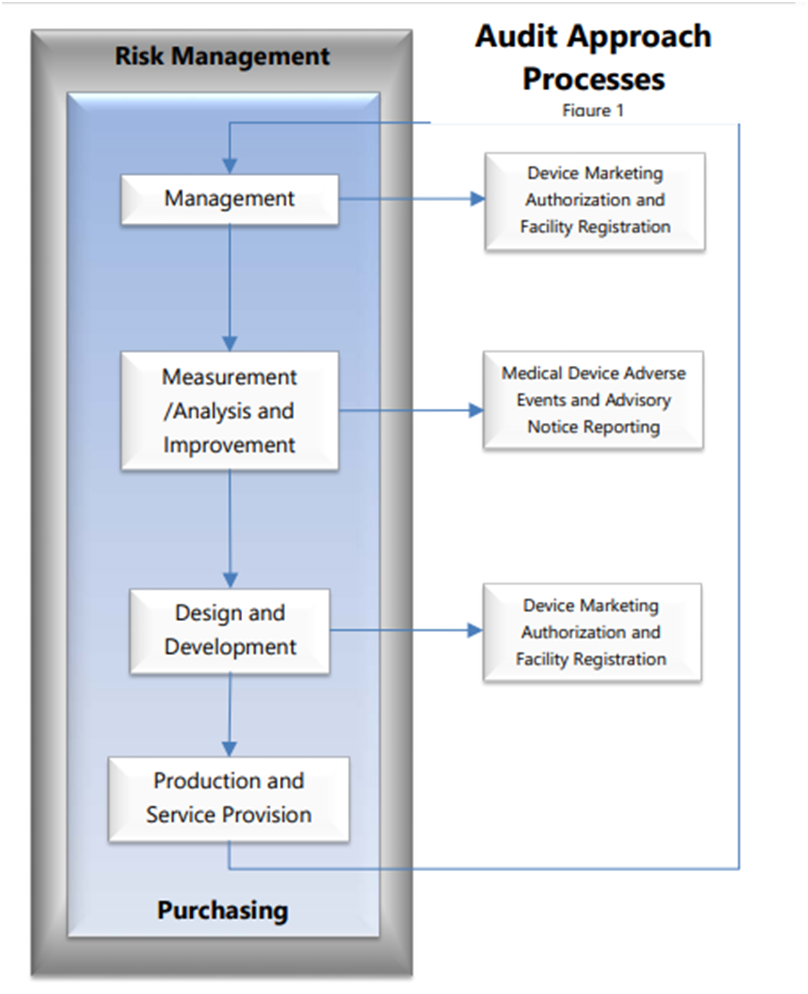
In today’s world of healthcare, technology plays an important role in the life cycle of medical devices and software has become an integral part of majority of products that serve both medical and non medical purposes.
There are three types of medical devices related to software. Namely, Software as a Medical Device (SaMD), Software in a medical device and software used in the manufacture or maintenance of a medical device.
Use of SaMD is increasing gradually. It can be used across a wide range of technological platforms such as medical device platforms, commercial “off-the-shelf” platforms, and virtual networks to name a few. Previously these were referred by industry, international regulators, and health care providers as “standalone software,” “medical device software,” and/or “health software,” and can sometimes be confused with other types of software.
Definition:
The definition of SaMD as defined by IMDRF is as follows “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”
Since the software possesses unique features which extend beyond the traditional medical device, there is a need to come up with a common framework and principles that enables all the stakeholders to promote innovation which is safe and protects patient safety. By keeping this in mind, International Medical Device Regulators Forum (IMDRF) in 2013 formed a group known as Software as a Medical Device Working Group (WG) aimed to develop guidance supporting innovation and timely access to safe and effective SaMD globally. The group mainly works on key definitions of SaMD, risk categorization, QMS and clinical evaluation of SaMD.
Following are the guidelines released by IMDRF to outline the key considerations of SaMD.
| Guidelines | Topics Covered |
| IMDRF/SaMD WG/N10 | Key Definitions |
| IMDRF/SaMD WG/N12 | Possible framework for Risk Categorization and corresponding considerations |
| IMDRF/SaMD WG/N23 | Application of QMS |
| IMDRF/SaMD WG/N41 | Clinical Evaluation |
Consideration as SaMD:
Software that is embedded as part of a hardware medical device and is necessary to drive the intended medical purpose IS NOT a SaMD. However, if the software is interfaced with other hardware medical devices and acts as an additional enhancement for its medical purposes can be considered as a SaMD.
Fitness or wellness apps are not considered as SaMD. It is clearly mentioned in both EU MDR 2017/745 and 520(o) (1) (A) – (D) of the FD&C Act (FDA) that are endorsed by IMDRF. In simple terms, a fitness or wellness software that are not related to diagnosis, cure, mitigation, prevention or treatment of a disease is not qualified to be called as SaMD.
The examples of “Not a SaMD software” are given below:
a) Mobile apps intended to manage stress
b) Mobile apps related to general lifestyle
c) Mobile apps that track baby’s sleeping and feeding habits
d) Apps that track sleeping patterns
Categorization of SaMD:
There are four risk categories identified based on the impact the SaMD has on the patient or public health.
Classification:
Unfortunately, different regulatory agencies have different criteria for classification.
United States:
The SaMD classification criteria used by USFDA are as same as traditional medical devices. Additionally, SaMD involves level of concern. While it may be strongly correlated with risk class, but level of concern is not used to determine your device’s risk classification.
EU:
There is no specific class for SaMD in EU. However, Medical device software uses the same risk classification as traditional medical devices: class I, class IIa, class IIb, and class III.
Rule 11 of EUMDR states:
Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
a) Death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or
b) A serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
IMDRF:
The IMDRF has put forth a guidance document to categorize SaMD. Based on the information provided in the document, there are four categories and the categorization is based on two factors namely, state of healthcare situation and significance of the information provided by the SaMD. The categorization table is provided below.
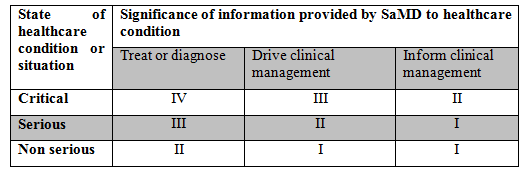
However, the IMDRF categorization is not widely used. But, it is helpful in determining the risk class in EU. One of the EU document, MDCG 2019-11, includes a table that combines both the IMDRF categorization and EU risk classes.
IEC 62304:
The IEC 62304 classification system has three levels based on the severity of injury that a software failure could cause:
-Class A – no injury
-Class B – non-serious injury
-Class C – serious injury or death
Challenges in regulating SaMD:
-Software acting as medical device makes the data accessible over network. By keeping this in mind, it is important to maintain the data safety while aligning the functionality and efficiency of the product.
-With the diversification of software in the healthcare, the security concerns are high. Stakeholders, regulators, and various authorities at multiple levels must comply with regulations set by the FDA to fast track approval of the Software being developed for healthcare applications.
-Due to the dynamic evolve of internet there is a need to balance innovation and adaptability without putting the user’s data at risk.
SaMD is a growing sector and with new parties entering the domain, new ideas are pouring in. Developers need to Developers need to balance innovation and regulatory compliance to develop smart healthcare without compromising data sensitive to user.
KN CONSULTING AND SERVICES is a well-oiled marvel in the field of Medical devices regulatory consulting. We can provide you with flexible and detailed planning and consulting services suiting to your needs.